Welcome to SittuAI Your Autonomous Regulatory AI Agent
Not another chatbot. SittuAI is an autonomous AI agent purpose-built for FDA 510(k) submissions — handling predicate analysis, document generation, and compliance validation end-to-end. Months of regulatory work, compressed into days.
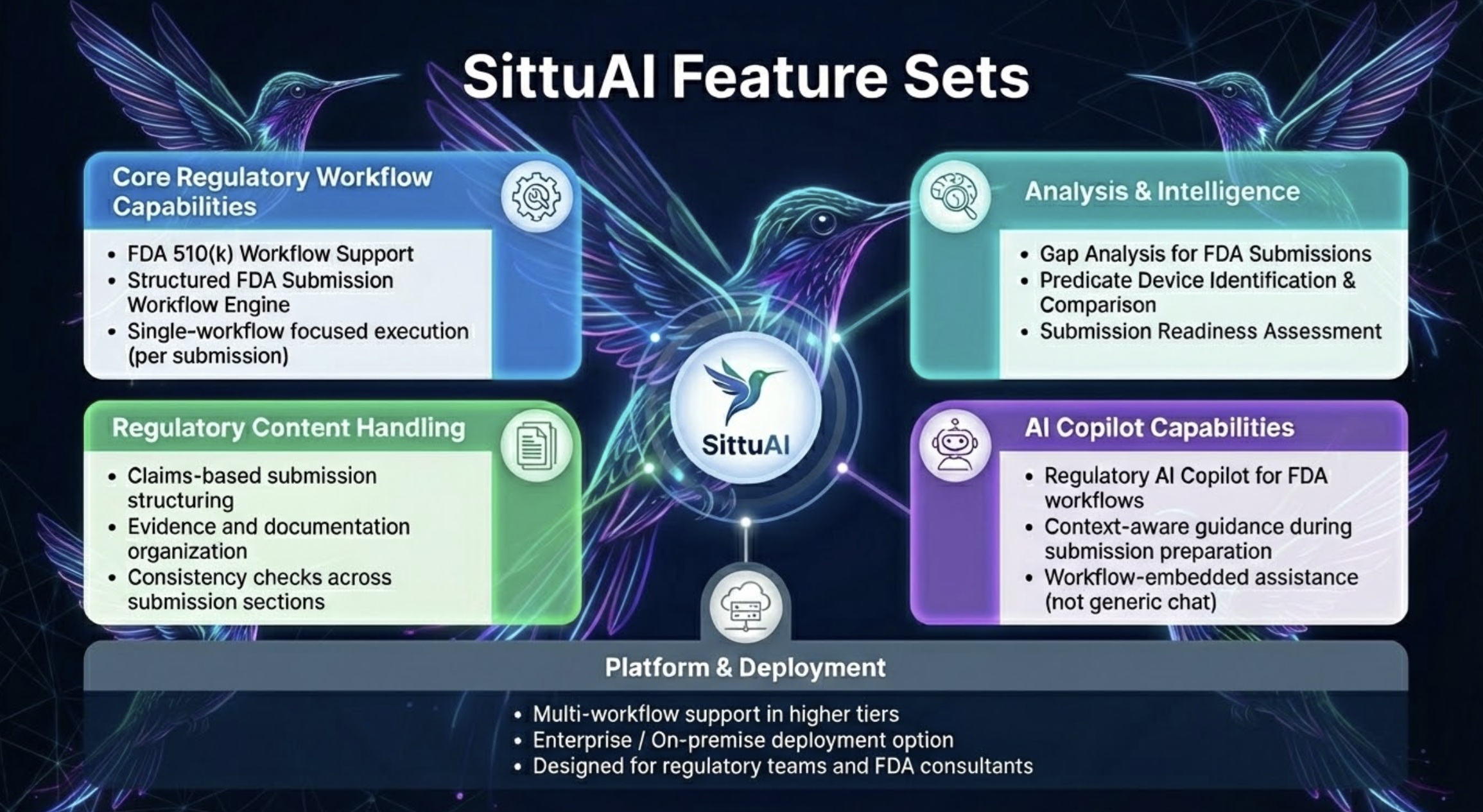
What Your AI Agent Does
Purpose-built AI grounded in 500K+ FDA predicate records, 21 CFR Part 820, and current FDA guidance
Predicate Intelligence
Instantly finds and analyzes similar cleared devices from our comprehensive FDA database.
SittuAI Chat
Ask anything about your submission. Get precise, FDA-grounded answers anchored to your device profile and 510(k) precedents — not generic AI responses.
AI Document Generation
Automatically generates 24+ compliant regulatory documents tailored to your device profile and FDA requirements.
Knowledge Graph
Connected intelligence that understands device relationships, regulations, and FDA guidance documents.
Compliance Validation
Real-time validation against 21 CFR requirements and current FDA guidance documents.
Simple 4-Step Process
From device profile to FDA-ready submission package
Define Your Device
7-step guided wizard
AI Analysis
Find predicates & gaps
Generate Documents
24+ docs auto-drafted
Export Package
eSTAR-ready format
From Device Profile to FDA-Ready Submission
A purpose-built platform that guides medical device companies through every step of the 510(k) journey — intelligently, efficiently, and with confidence.
Intelligent Device Setup
Get started in minutes with our guided 7-step device onboarding wizard. Define your device's intended use, classification, target users, and predicate strategy. Our AI builds a rich Device Knowledge Graph that powers every downstream workflow — from document generation to predicate matching.
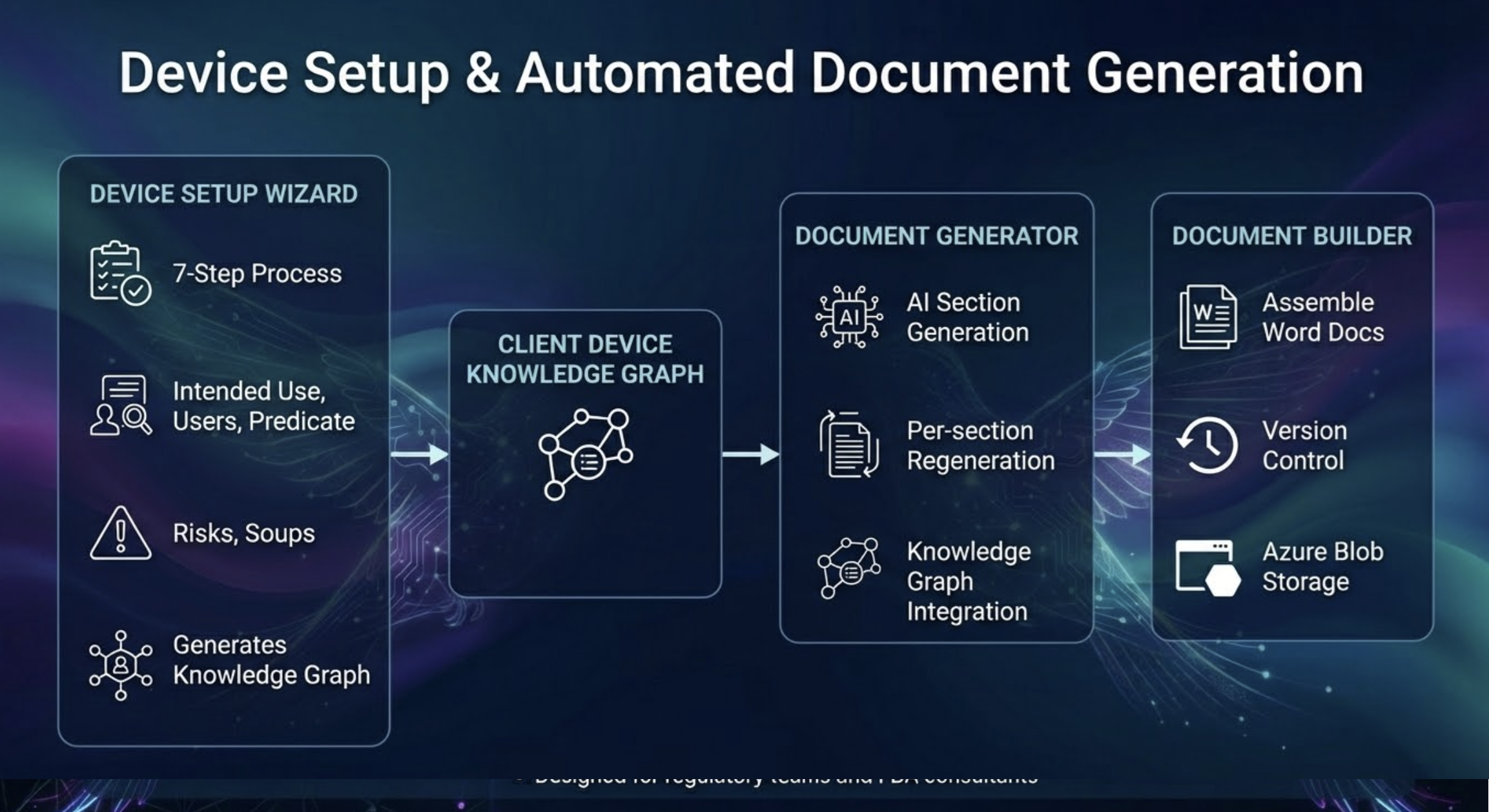
Structured Regulatory Workflow
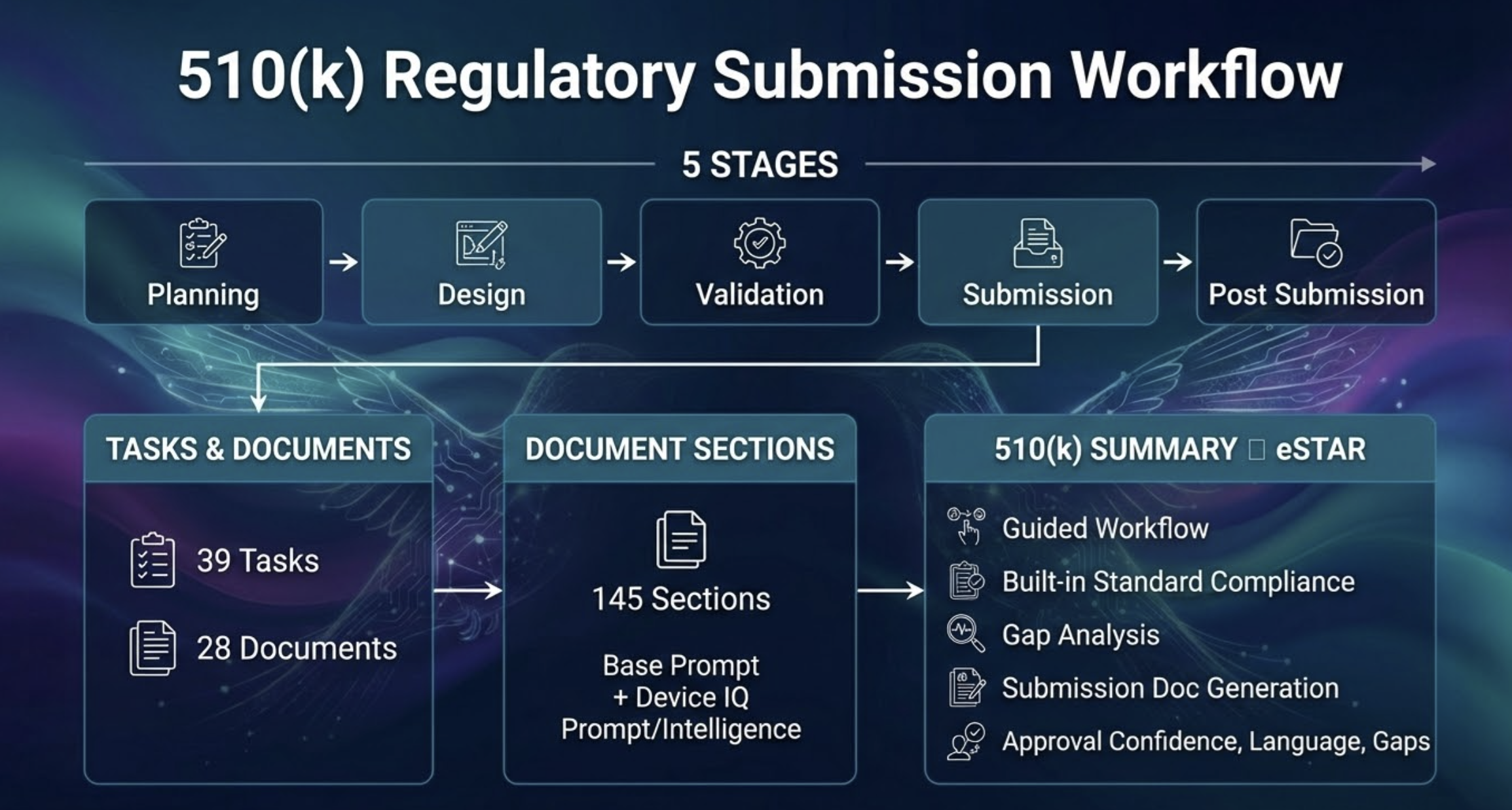
Follow a proven, stage-gated submission workflow built around FDA expectations. SittuAI breaks the 510(k) journey into clear phases so your team always knows what's next, what's pending, and what's complete — with full progress visibility across every stage.
AI-Powered Document Generation
Generate all 24+ regulatory documents from a single device profile. Each document comes with section-level tracking, individual approval workflows, and AI-assisted content generation per section — so your team can review, edit, and approve with precision rather than starting from scratch.
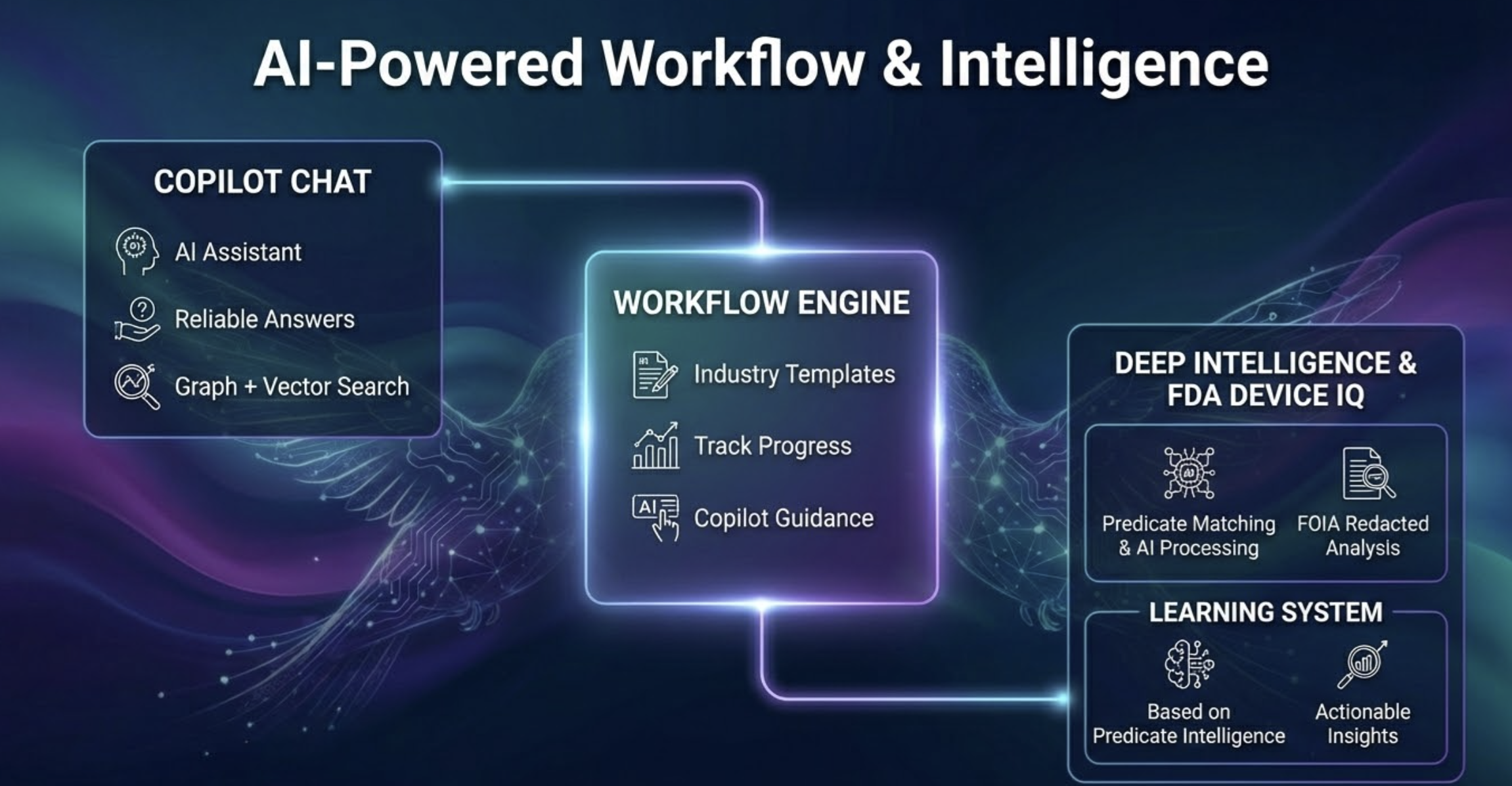
SittuAI Chat
Ask anything. Get precise, grounded answers. SittuAI Chat is trained on FDA guidance documents, 510(k) precedents, and your device's own knowledge graph — delivering consistent, reliable answers specific to your submission rather than generic AI responses.
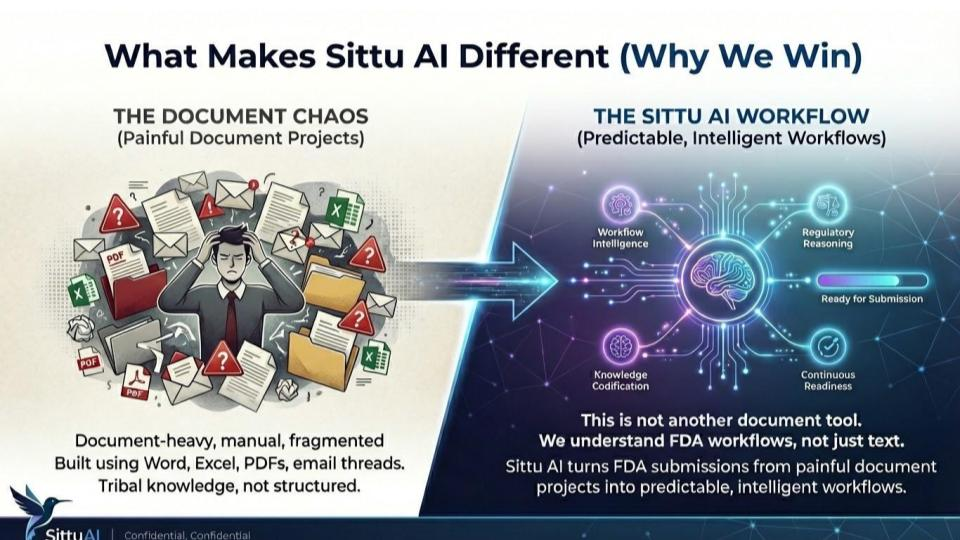
The SittuAI Advantage
Unlike generic AI tools, SittuAI is purpose-built for FDA 510(k) submissions. Every feature — from predicate intelligence to document assembly — is designed around real regulatory workflows. The result: up to 60% faster submission preparation, fewer revision cycles, and teams that spend time on strategy, not paperwork.
Ready to Transform Your 510(k) Submission?
Join medical device companies who are submitting faster, smarter, and with confidence.
Grow your consulting practice — without growing the grind.
80% of your time is spent on documentation, not expertise. SittuAI handles the drafting work so you can focus on what clients actually pay you for: validation, strategy, and FDA know-how. Join our platform for free and get matched with medical device companies who need your expertise.
Serve more clients, faster
AI drafts the documents needed per submission. You validate, not rewrite. Take on 3-5× more clients in the same time.
Get matched with new customers
Medical device, Pharma & Cosmetic companies and SaMD startups come to us for help. We route qualified clients directly to consultants on our platform.
Free to join.
No subscription, no platform fees. Use SittuAI's AI-powered Predicate Matching & Co Pilot chat for your own client engagements.
Cut submission time in half
Validated by a working FDA consultant: 60% reduction in total 510(k) submission time. Deliver faster, impress clients, win more repeat business.
Your expertise stays yours
You validate every document. Your name, your credentials, your sign-off. SittuAI is the leverage — you're still the expert.
510(k), De Novo, PMA ready
Pre-built workflow templates for every major FDA submission pathway. Knowledge graph built from FDA regulations, guidance, and real precedents.
Takes 2 minutes. We'll be in touch within 1 business day.
Latest FDA Regulatory Insights
Expert-written guides on 510(k), De Novo, PMA submissions, and compliance strategy.
Biocompatibility Testing for 510(k): A Complete Submission Guide
Biocompatibility testing for 510(k) submissions requires a risk-based evaluation framework per FDA's 2016 ISO 10993-1 guidance, not a fixed checklist—mistakes in scope, standard application, or data presentation commonly cause 3-6 month delays. Success depends on a robust Biological Evaluation Plan and Report that justify endpoint selection based on device contact type, duration, materials, and lifecycle factors.
How FDA Classifies Medical Devices: Class I, II, and III Explained
**Summary:** FDA classifies medical devices into three risk-based categories that determine the regulatory pathway (510(k), De Novo, or PMA) and oversight required. Correct classification is critical—wrong classification can delay market entry by 12–18 months or trigger enforcement action.
Real-World Evidence for Medical Device Regulatory Decisions: An Interpretive Guide
The FDA now accepts real-world evidence (RWE)—data from clinical practice outside controlled trials—to support device approvals, expansions, and postmarket requirements under an evolving regulatory framework. Companies must ensure their real-world data is fit for purpose, methodologically rigorous, and properly documented to meet FDA's credibility standards across the device lifecycle.
Ready to Transform Your Regulatory Submissions?
Join leading medical device companies who have reduced their 510(k) preparation time by 60%.